
domestica stocks and made use of ChIP seq to carry out the 1st ab initio look for putative gene pro moters that are concurrently marked by mutually exclu sive, transcriptionally opposing histone modifications being a indicates to identify candidate imprinted genes. Success ChIP seq analysis The genomic distributions of four histone modifications were analyzed in opossum fibroblasts by ChIP seq, using antibodies towards H3K4me3, H3K9me3, H3K27me3, and H3K9Ac, A lot more than 436 million Illumina ChIP seq reads from male fibroblasts were uniquely mapped for the recent M. domestica genome assembly, The two marks of activation exa mined, H3K4me3 and H3K9Ac, gave 79,412 and 52,511 exceptional peaks of enrichment, respectively, The two marks of repression examined, H3K9me3 and H3K27me3, gave 56,719 and sixteen,592 unique peaks of en richment, respectively, We subsequent analyzed the overlap of each histone modification with promoters of annotated genes and their linked CpG islands.
Within the 22,030 annotated genes in MonDom5, 13,021 showed expression in a minimum of one of 4 male fibroblast cell lines as determined by RNA seq, and 9,012 of them had been marked by H3K4me3, About half of those expressed genes selleck chemicals HDAC Inhibitor have an annotated CpG island on the professional moter and 93% of these CpG islands were marked with H3K4me3 irrespective of transcriptional state, As a result, the promoters of your transcribed genes showed enrichment for two MOAs and were deficient for MORs, whereas the promoters of repressed genes showed a deficiency in MOAs and, in some instances, an enrichment of H3K9me3. The dis tribution of H3K27me3 was diffuse across the opossum genome. Most major peaks occurred in intergenic re gions, although promoters and gene bodies of biallelically expressed genes and identified opossum imprinted genes showed a standard depletion of H3K27me3.
On top of that, en richment selleck chemicals of H3K27me3 hasn’t been shown in other mammalian species to become mutually exclusive using the MOAs used in this study. For these good reasons H3K27me3 appeared not to be valuable for the functions of this study and was excluded from additional examination. Along with the promoters discussed over, we exam ined overlap of your a variety of histone modifications with each other and all annotated putative promoters within the MonDom5 assembly. With the H3K9Ac peaks, 47,275 overlapped with an H3K4me3 peak by at the very least one base pair, and 6,410 H3K9me3 peaks overlapped with an H3K4me3 peak, Also, 11,580 promoter related CpG islands were marked by a substantial H3K4me3 peak. In the 35,105 putative promoters, sixteen,620 were marked  with H3K4me3, 7,871 also had an annotated CpG island, and 179 of them have been also concurrently marked with H3K9Ac and H3K9me3, No X linked genes met these criteria. This really is noteworthy be cause the fibroblasts analyzed were of male origin, and as a result possessed only a single X chro mosome.
with H3K4me3, 7,871 also had an annotated CpG island, and 179 of them have been also concurrently marked with H3K9Ac and H3K9me3, No X linked genes met these criteria. This really is noteworthy be cause the fibroblasts analyzed were of male origin, and as a result possessed only a single X chro mosome.
domestica stocks and used ChIP seq to complete the very first ab
Reply
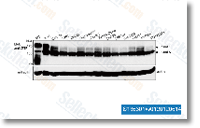 siRNA for 3 consec utive days we were not capable to cut back the amounts of PKCcompletely which raises the probability that much more suppressive results might be obtained if PKCcould be depleted from the cells. A purpose of PKCis in line with the suppression on the TPA result obtained by the standard PKC inhibitor GF109203X. Nevertheless, in contrast to PKCsiRNA remedy, the kinase inhibitor didn’t affect migration under basal ailments. PKChas been shown to induce morphological effects, induction of neurites and dismantling of anxiety fibres. independently of its kinase activity. Our effects indicate that also a lot of the promigratory effects of PKCmay be exerted inde pendently of its catalytic exercise. The inhibitor of classical PKCs, G6976, also suppressed migration, indicating a potential part for these isoforms in migration. Even so, G6976 influenced migration the two inside the absence and presence of TPA contrasting the impact of GF109203X, which didn’t have an result underneath basal conditions.
siRNA for 3 consec utive days we were not capable to cut back the amounts of PKCcompletely which raises the probability that much more suppressive results might be obtained if PKCcould be depleted from the cells. A purpose of PKCis in line with the suppression on the TPA result obtained by the standard PKC inhibitor GF109203X. Nevertheless, in contrast to PKCsiRNA remedy, the kinase inhibitor didn’t affect migration under basal ailments. PKChas been shown to induce morphological effects, induction of neurites and dismantling of anxiety fibres. independently of its kinase activity. Our effects indicate that also a lot of the promigratory effects of PKCmay be exerted inde pendently of its catalytic exercise. The inhibitor of classical PKCs, G6976, also suppressed migration, indicating a potential part for these isoforms in migration. Even so, G6976 influenced migration the two inside the absence and presence of TPA contrasting the impact of GF109203X, which didn’t have an result underneath basal conditions.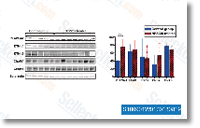 formation of water canal openings inside the exopinaco derm, Molecules while in the Notch and TGF B signal ing pathways had been also detected in all phases, despite the fact that the expression of ligands and receptors were not usually coordinated.
formation of water canal openings inside the exopinaco derm, Molecules while in the Notch and TGF B signal ing pathways had been also detected in all phases, despite the fact that the expression of ligands and receptors were not usually coordinated.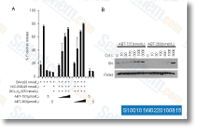 had been down regulated in MT in contrast with WT, These integrated the genes encoding a NADH ubiquinone oxidor eductase 75kDa subunit, cytochrome C oxidase, and cytochrome oxidase subunit 3, suggesting that MT mitochrondria have been capable of less efficient electron transport than that WT ones. If, like a outcome, flux by means of the TCA cycle is decreased, the accu mulation of citric acid is more likely to be compromised. Furthermore, PDS and ZDS catalyzing desaturation of phy toene to lycopene involve net electron transfer, In tomato, a NAD H dehydrogenase complex which parti cipating in electron transfer was concerned in carotenoid biosynthetic pathway, suggesting the doable exist of cross talk amongst electron transfer and carotenoid accu mulation in sweet orange.
had been down regulated in MT in contrast with WT, These integrated the genes encoding a NADH ubiquinone oxidor eductase 75kDa subunit, cytochrome C oxidase, and cytochrome oxidase subunit 3, suggesting that MT mitochrondria have been capable of less efficient electron transport than that WT ones. If, like a outcome, flux by means of the TCA cycle is decreased, the accu mulation of citric acid is more likely to be compromised. Furthermore, PDS and ZDS catalyzing desaturation of phy toene to lycopene involve net electron transfer, In tomato, a NAD H dehydrogenase complex which parti cipating in electron transfer was concerned in carotenoid biosynthetic pathway, suggesting the doable exist of cross talk amongst electron transfer and carotenoid accu mulation in sweet orange. were studied by our crew. As a way to comprehend the impact of JA and MeJA on L. gmelinii on the transcriptional level, the examination of differentially expressed genes making use of digital gene expression was carried out. The differential gene expression profiles could possibly deliver an invaluable re source for the investigation of molecular mechanisms in L. gmelinii disease/insect resistance and their prospective defensive signals.
were studied by our crew. As a way to comprehend the impact of JA and MeJA on L. gmelinii on the transcriptional level, the examination of differentially expressed genes making use of digital gene expression was carried out. The differential gene expression profiles could possibly deliver an invaluable re source for the investigation of molecular mechanisms in L. gmelinii disease/insect resistance and their prospective defensive signals.